
 Breast Cancer
Breast Cancer
Approximately 1 in 8 women will have a diagnosis of breast cancer over their lifetime. This case looks at what makes breast tissue so susceptible to cancer and examines particular risks, both environmental and genetic, that contribute to this high incidence. Breast epithelial cells receive growth signals via hormones as part of a normal menstrual cycle. Compared to cells in other tissues, they reproduce at a relatively high rate. This permits somatic mutations in breast tissue cells to occur and accumulate at a high rate, leaving breast tissue prone to cancer. Some germ line (inherited) mutations also contribute to the onset of breast cancer. The persistence of these mutations can potentially be explained as an evolutionary trade-off or via founder effects. In other cases, the onset of breast cancer can be explained by exposure to environmental factors, such as modern diets, cigarettes, and changes in reproductive patterns.
This case will introduce the mechanistic and evolutionary underpinnings of breast cancer. It will also explore how an evolutionary perspective can be utilized to improve the treatment of some cases of breast cancer.
Multicellular organization and cancer
Cells are the smallest individual units of life. The evolution of multicellularity allowed for the division of labor (e.g. movement, digestion, reproduction etc) between different cell/tissue types. However, multicellular life also has an inherent risk of cancer: Under certain conditions, individual cells may be selected to selfishly replicate at the expense of the organism. Cancers result from an accumulation of genetic mutations and can disrupt the regulatory processes within a cell cycle. Cells become self-interested, dividing on their own schedule, out of sync with others that surround them. They “cheat” the cellular system in which they reside, acting more like single celled organisms. They often start to look different from other cells as well. They outpace the reproduction of normal cells and can form a mass called a neoplasm. A neoplasm can progress to become tumor. Some tumors are harmless, benign, while others are malignant or cancerous. The progression from a harmless to cancerous tumor primarily depends on the function of enzymes and proteins that 1) regulate cell growth and division or 2) ensure faithful apoptosis and DNA synthesis. In breast cancer, the former includes protein receptors for epidermal growth factor (HER2) and the latter includes proteins involved in apoptosis (TP53) and DNA repair (BRCA2). The three genes corresponding to these proteins are discussed at some length in the Genetics module.
DNA synthesis, mitosis, and the HER2 & ESR1 proteins
Living cells need to occasionally replicate in order to keep tissues functioning and healthy. Cellular replication involves three major processes: DNA replication, mitosis, and cytokinesis. Before a cell can replicate, it must first synthesize new DNA. During this process, the strands of the double helix separate, each becoming a template for synthesis of a new strand. This is considered semi-conservative replication, since each progeny strand is directly derived from the parent molecule. During the synthesis of new DNA, mistakes in the nucleotide base sequence (i.e. mutations, discussed above) can occur. Sometimes these mistakes are dealt with via apoptosis or DNA repair (discussed below), but often they become part of the next generation of cells in our body. In fact, all of the somatic cells in adult bodies represent a “patchwork” of various mutations, sometimes referred to as somatic mosaicism. It is estimated that most tumors contain around 1000 to 20000 point mutations and hundreds of insertions, deletions, and rearrangements (Martincorena & Campbell, 2015).

There are a variety of proteins that are expressed in order to promote rapid cellular growth and replication. In some cases, this process is necessary to sustain healthy tissues (e.g. when there is damage). However, when these proteins are overexpressed, cells can begin to replicate more than they need to. For example, when an extracellular growth factor binds to a growth factor receptor (a protein product of HER2), a series of events leads to the promotion of cellular growth and replication. Therefore, any genetic change that leads to an overexpression of these membrane receptors can increase the rate of cell proliferation and ultimately lead to cancer.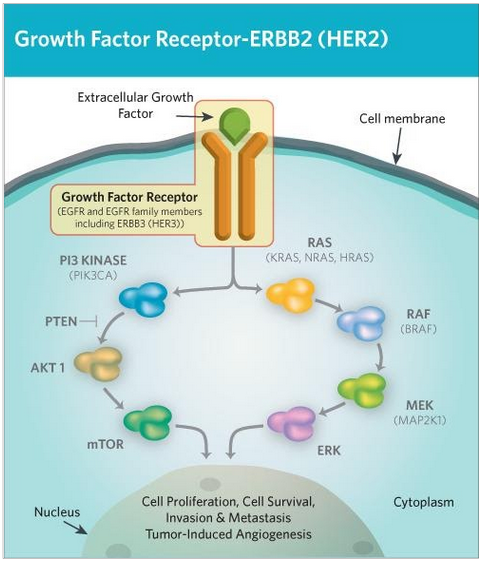
ESR1 is an estrogen receptor that consists of 595 amino acids. When bound to estrogen, travels to the nucleus where it increases the transcription of many genes that increase the rate of cell growth and replication. It is expressed in cells throughout the body, but it specifically fosters cell reproduction associated with the menstrual cycle, including the growth of breast tissue. The differences in estrogen levels between the sexes partially contributes to explain the increased risk of breast cancer in women compared to men.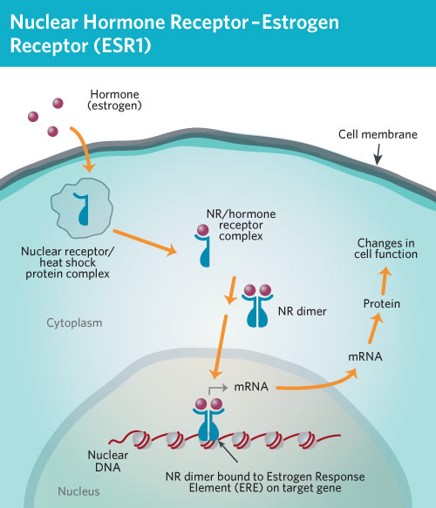
Apoptosis and the TP53 protein
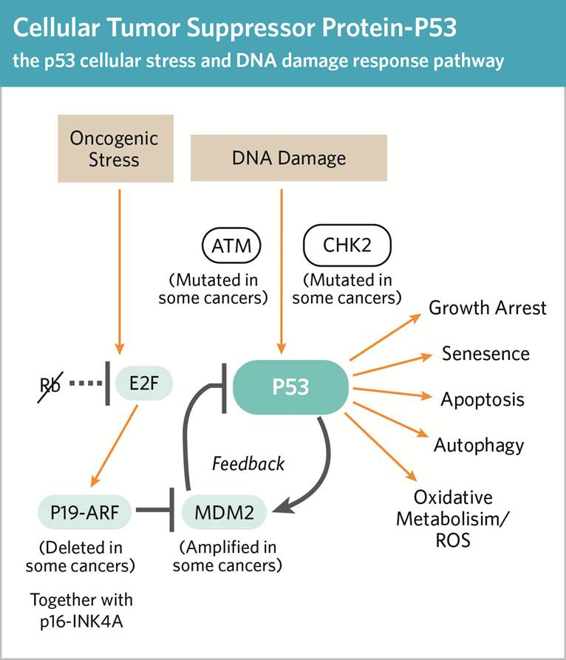
Apoptosis is cell self-destruction that happens often during tissue development. Apoptosis is an important cellular process in the body as it gets rid of cells that have the potential to become cancerous (e.g. cells with numerous mutations and/or damaged DNA). Triggers can be external messages that bind to corresponding cell receptors on the cell membrane or by internal messages, generated by stress (eg heat, radiation, starvation, etc). One internal stressor is accumulation of errors in DNA synthesis sensed by checkpoint proteins such as p53 (the protein product of TP53). Once a cell gets and processes the signal to self-destruct, it undergoes a series of biochemical and morphological changes.

- A group of enzymes, caspases, activate the destruction of cellular organelles and the degradation of mRNA
- The cell shrinks as the cytoskeleton collapses: cell contents crowd together.
- The cell membrane folds in on itself and loses its structural integrity.
- In the nucleus, chromatin clumps form as DNA fragments.
- The cell collapses in on itself.
- The cell breaks apart, forming apoptotic fragments, which are cleared out by special cells called phagocytes.
There are both pro-apoptotic and anti-apoptotic proteins that regulate its occurrence. For example, p53 is a key pro-apoptotic factor. When released to the cytoplasm, p53 activates the cascade of caspases that lead to cell suicide. Other proteins, such as MDM2 inhibit pathways leading to cell death. Overexpression of these proteins can affect cell self-destruction by suppressing p53 and subsequent caspase cascades. Cancer is often associated with an increase in the pro- compared to the anti-apoptotic proteins. This imbalance allows these cells to proliferate, carrying their stressors and errors to the next generation of cells.
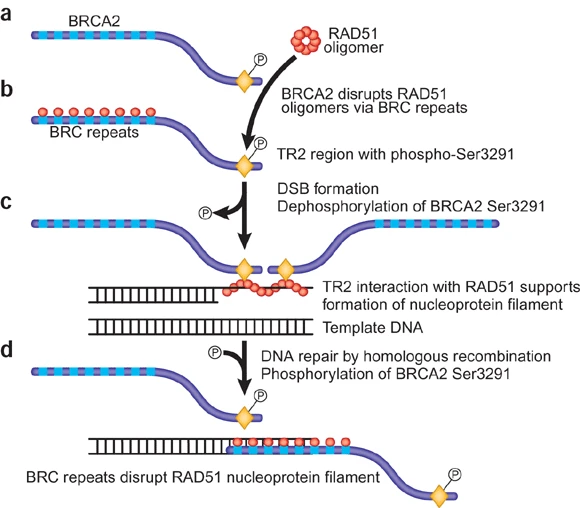
DNA Repair and the BRCA2 Protein
The evolutionary puzzle of cancer: Why does breast cancer persist?
If breast cancer can often result in early mortality, then why is it so common throughout human populations? If natural selection tends to lead to increased fitness of the individuals within populations over time, then wouldn’t we expect harmful gene variants such as dysfunctional copies of BRCA1/2 to be selected out of the population? These questions can be addressed by two broad concepts: Firstly, natural selection is only one of five evolutionary mechanisms. Along with selection, these other four mechanisms (mutation, drift, gene flow, and sexual selection), are also constantly impacting the frequency of alleles within populations. Secondly, natural selection is often constrained by a variety of factors. For example, rapidly changing environments can often outpace natural selection. The sections below discuss how these evolutionary mechanisms and constraints can lead to the prevalence of breast cancer within human populations.
Antagonistic pleiotropy and trade-offs
An evolutionary trade-off refers to a situation in which evolution cannot optimize the fitness associated with a specific trait without compromising the fitness associated with another. One common source of trade-offs is antagonistic pleiotropy. Pleiotropy refers to when a single gene influences two or more phenotypic traits, and antagonistic refers to the fact that these two phenotypes oppose each other in terms of their effects on an individual’s evolutionary fitness (i.e. one positive effect, and one negative effect). Antagonistic pleiotropy usually occurs in a life-history context, where a gene variant that has positive fitness effect early in life (i.e. leading up to or during a reproductive age) has a negative effect later in life (i.e. post-reproductive age). Natural selection often favors these variants because they improve fitness during a reproductive age, even though there a cost later in life (i.e. a trade-off).
There is some evidence to suggest that BRCA1/2 variants in humans represents a case of antagonistic pleiotropy, where the same variants that increase breast cancer risk also appear to be linked to increased reproductive success. Specifically, historical family data has uncovered trends in various metrics of fertility between carriers and non-carriers of BRCA1/2 variants. Smith et al. (2011) reported that women with BRCA1/2 mutations tended to bear more offspring over their lifetime compared to women without BRCA1/2 mutations, but they also had an increased risk of mortality at a post-reproductive age. Mutation carriers also tended to have shorter intervals between births and ended childbearing at a later age than controls. In a similar study, Kwiatkowski et al. (2015) report that BRCA 1/2 mutation carriers were less likely to bear no children in their lifetime, and tended to have more children overall. Although these data are intriguing, it is worth noting that contradictory evidence suggests these trends may not be generalizable across populations.

Founder effects and population genetics

Environmental mismatches
A final explanation for high rates of breast cancer in human populations is that we live in vastly different environments compared to a majority of ancestral humans. Natural selection is a process that occurs over many generations, but human environments can change rapidly, especially since the recent industrial and technological revolutions. In general, modern humans have different diets, reproductive patterns, and lifestyles compared to ancestral humans. The asymmetry between ancestral and modern conditions is called an environmental mismatch.
One element that differs quite dramatically between many modern and ancestral human populations are typical reproductive patterns. In many modern cultures, birth rates have drastically declined. In the USA, the average woman has over 400 menstrual cycles compared to around 100 in a culture in Africa without birth control and lower rates of breast cancer (Strassmann, 1999). The mechanism behind this is likely related to increased hormone exposure, as estrogen receptors in breast tissue initiate cell division, and exposure to compounds that mimic estrogens have been implicated in higher rates of breast cancer. As a result, breast cancer estimated to be 10x higher in women in the USA compared to hunter-gatherers (Eaton et al., 1994). Furthermore, a meta-analysis identified the number of pregnancies as being a significant factor associated with reduced risk of breast cancer (Ewertz et al., 1990). Finally, across cultures, there is a strong association between breast cancer risk and average levels of progesterone during menstruation (Jasienska & Thune, 2001), suggesting a relationship between lifestyle factors, hormones, and the risk of breast cancer.
Aside from reproductive patterns, there are many other elements of typical modern environments that contribute to increased rates of breast cancer. For example, about one third of all cancer can be attributed in part to tobacco use, and another third to obesity. Another major risk factor associated with many cancers is age. On average, humans in modern environments have longer lifespans, meaning the risk of cancer within one’s lifetime is also higher.